
Blog
Is PSSM2 Genetic?
13 Dec 2019

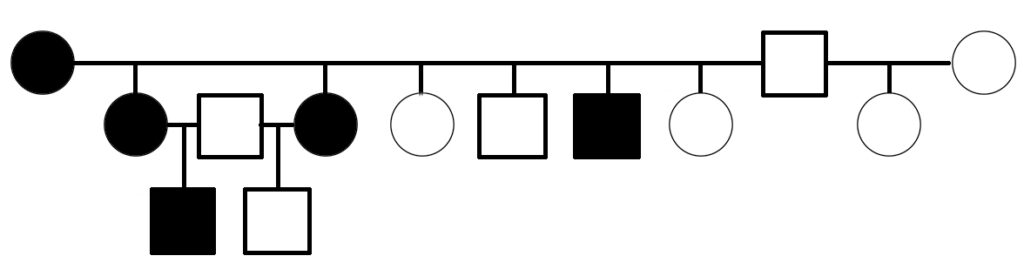
Is PSSM2 genetic? By genetic, we mean that it is an inherited condition, transmitted from parents to offspring rather than resulting exclusively from environmental conditions, such as diet or exposure to toxins or pathogens. What do we mean by PSSM2?
PSSM2 is an abbreviation for Polysaccharide Storage Myopathy type 2 [1]. It is a disease state in horses initially defined by the following criteria: symptoms of exercise intolerance, a negative DNA test for GYS1-R309H (a genetic variant of the gene encoding glycogen synthase that is associated with the disease state PSSM1), and abnormalities observed in muscle biopsy [1].
The first evidence suggesting that PSSM2 is genetic was presented in 2007, when Dr. Molly McCue presented a pedigree of affected Quarter Horses related by descent from a common ancestor in her PhD dissertation. Related horses were diagnosed with PSSM2 by muscle biopsy.
Polysaccharide Storage Myopathy type 1 (PSSM1) was shown to be associated with GYS1-R309H in 2008 [2]. The GYS1-R309H variant has been shown to encode a constitutively activated glycogen synthase [3]. The improper regulation of this enzyme pushes the equilibrium between glycogen synthesis and degradation in the direction of glycogen synthesis, even when the affected horse’s energy needs would normally favor the breakdown of glycogen to provide glucose for energy.
In contrast, genome-wide association studies (GWAS) on horses diagnosed with PSSM2 have failed to identify a gene responsible for all cases of this disease state. In addition, whole genome sequencing of horses diagnosed with PSSM2 has failed to identify a particular genetic variant among candidate genes that is consistently associated with PSSM2 [4].
A subtype of PSSM2, Myofibrillar Myopathy (MFM), was first described in Arabian horses [5]. In Arabian MFM, horses show symptoms of exercise intolerance, test negative for GYS1-R309H, and show abnormalities on muscle biopsy. In addition, muscle biopsies from horses with Arabian MFM show desmin-positive aggregates outside of the Z disc, and electron microscopy shows breaks in the Z disc and Z disc streaming. Arabian MFM presents the false appearance of a glycogen storage disorder, because pools of glycogen granules of normal size form in areas where the structure of myofibrils is disorganized [5].
A different subtype of MFM has been described in Warmblood horses [6]. Warmblood MFM differs from Arabian MFM in that while Arabian MFM is associated with an exercise-induced elevation of serum creatine kinase (CK), Warmblood MFM is not. Like horses with Arabian MFM, horses with Warmblood MFM show desmin-positive aggregates outside of the Z disc by immunostaining, and breaks in the Z disc and Z disc streaming by electron microscopy [6].
Warmblood MFM has been shown to be genetic. A three-generation pedigree shows horses affected by Warmblood MFM, as scored by muscle biopsy, including electron microscopy [6]. The pedigree suggests that Warmblood MFM is inherited as a dominant or semidominant trait.
In human patients, Myofibrillar Myopathy has been associated with mutations in nine different genes: DES [7], CRYAB [8], MYOT [9], LDB3 [10], FLNC [11], BAG3 [12], KY [13], PYROXD1 [14], and TTN [15]. There are also cases of human MFM that lack a molecular diagnosis, suggesting that variants of additional genes are also associated with MFM. Human patients with MFM have similar symptoms, and the causative genetic variant cannot be identified without DNA testing. No human cases of MFM have been associated with environmental factors like diet or exposure to toxins or pathogens.
It is a fundamental premise of genomics and molecular genetics that mammals share a common set of genes derived by descent from a common ancestor, and that investigations of inherited conditions in one mammal are of value in understanding similar conditions in other mammals. It is very likely that PSSM2 and MFM in horses resemble the related disease states in human patients, where they are known to be genetic.
Citations
[1] McCue, ME et al. (2009). “Comparative skeletal muscle histopathologic and ultrastructural features in two forms of polysaccharide storage myopathy in horses.” Vet Pathol. 46(6):1281-1291. PMID: 19605906.
[2] McCue, ME et al. (2008). “Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis.” Genomics. 91(5):458-466. PMID: 18358695.
[3] Maile CA et al. (2017). “A highly prevalent equine glycogen storage disease is explained by constitutive activation of a mutant glycogen synthase.” Biochim Biophys Acta Gen Subj. 1861(1 Pt A):3388-3398. PMID: 27592162.
[4] Valberg S (2019). Website of Stephanie Valberg, College of Veterinary Medicine, University of Michigan, viewed December 2019.
[5] Valberg S et al. (2016). “Suspected myofibrillar myopathy in Arabian horses with a history of exertional rhabdomyolysis.” Equine Vet J. 48(5):548-556. PMID: 26234161.
[6] Valberg S et al. (2017). “Clinical and histopathological features of myofibrillar myopathy in Warmblood horses.” Equine Vet J. 49(6):739–745. PMID: 28543538.
[7] Park KY et al. (2000). “Desmin splice variants causing cardiac and skeletal myopathy.” J Med Genet. 37(11):851-857. PMID: 11073539.
[8] Vicart P et al. (1998). “A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy.” Nature Genet. 20(1):92-95. PMID: 9731540.
[9] Gilchrist JM et al. (1988). “Clinical and genetic investigation in autosomal dominant limb-girdle muscular dystrophy.” Neurology. 38(1):5-9. PMID: 3275904.
[10] Selcen D and Engel AG (2005). “Mutations in ZASP define a novel form of muscular dystrophy in humans.” Ann Neurol. 57(2):269-276. PMID: 15668942.
[11] Vorgerd M et al. (2005). “A mutation in the dimerization domain of filamin C causes a novel type of autosomal dominant myofibrillar myopathy.” Am. J. Hum. Genet. 77(2):297-304. PMID: 15929027.
[12] Jaffer F et al. (2012). “BAG3 mutations: another cause of giant axonal neuropathy.” J Peripher Nerv Syst. 17(2):210-216. PMID: 22734908.
[13] Straussberg R et al. (2012). “Kyphoscoliosis peptidase (KY) mutation causes a novel congenital myopathy with core targetoid defects.” Acta Neuropathol. 132(3):475-478. PMID: 27484770.
[14] O’Grady GL et al. (2016). “Variants in the Oxidoreductase PYROXD1 Cause Early-Onset Myopathy with Internalized Nuclei and Myofibrillar Disorganization.” Am J Hum Genet. 99(5):1086-1105. PMID: 27745833.
[15] Pfeffer G et al. (2014). “Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure.” J Neurol Neurosurg Psychiatry. 85(3):331-338. PMID: 23486992.
Share this post
From the blog
The latest industry news, interviews, technologies, and resources.
P4 Allele of MYOZ3
Introduction This blog post explores the P4 allele (S42L) of the equine MYOZ3…
1 Apr 2025
P2 Allele of MYOT
Introduction This blog post explores the P2 allele (S232P) of the equine MYOT…
8 Mar 2025
EquiSeq is a biotech company offering
genetic testing for horses.
Resources
© 2017 – 2023 EquiSeq Inc. All rights reserved.