
Blog
What is PSSM?
30 Jan 2024

Polysaccharide Storage Myopathy (PSSM) is a form of equine exercise intolerance characterized by episodes of tying up. Horses with this condition have a particular set of clinical signs, including displays of pain, refusal to move forward, trouble standing for the farrier, and standing “parked out” as if to urinate. All of these are signs of a disease state affecting muscle.
Affected horses often show enlarged granules of glycogen in muscle tissue obtained by biopsy. These enlarged glycogen granules are relatively resistant to digestion by amylase, an enzyme that completely degrades normal glycogen granules under standard conditions.
Glycogen is a polysaccharide, a polymer of the simple sugar glucose, a monosaccharide. The synthesis of glycogen from glucose in muscle tissue provides a store of energy. When energy is needed, the glycogen is broken down into glucose, which can be metabolized for energy.
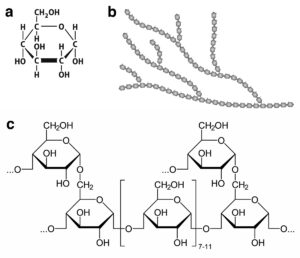
The image above shows a) the chemical structure of glucose, b) the general structure of glycogen in which glucose units are joined in a branching structure, and c) the chemical structure of the linear polymers of glucose and the branch points.
The synthesis of glycogen from glucose is highly regulated. In normal horses, glycogen is synthesized when energy needs are low compared to the amount of available glucose, and broken down back into glucose when energy is needed. Two enzymes are responsible for the synthesis of glycogen. One enzyme catalyzes the synthesis of the linear chains and the other catalyzes the synthesis of the branch points. Two different enzymes are responsible for the breakdown of glycogen. One cleaves the linkages in the linear parts of glycogen and the other catalyzes the cleavage of the branch points.
The finding of enlarged glycogen granules in muscle tissue of horses with PSSM suggested that the disease is a glycogen storage disease. This disease state has been described in human patients with defects in a number of different genes as shown in the table below.
| Disease state | Affected gene | Reference |
|---|---|---|
| GSD1a | G6PC | 1 |
| GSD2 | GAA | 2 |
| GSD3 | AGL | 3 |
| GSD4 | GBE1 | 4 |
| GSD5 | PYGM | 5 |
| GSD6 | PYGL | 6 |
| GSD7 | PFKM | 7 |
| GSD9 | PHKA2 | 8 |
| GSD10 | PGAM2 | 9 |
| GSD11 | LDHA | 10 |
| GSD12 | ALDOA | 11 |
| GSD13 | ENO3 | 12 |
| GSD14 | PGM1 | 13 |
| GSD15 | GYG1 | 14 |
Quarter Horses have a relatively high incidence of PSSM, a condition that seemed to be hereditary, as are the human glycogen storage diseases. A genome-wide association study showed that the presence of enlarged glycogen granules in muscle tissue was associated with a particular genetic variant of the gene encoding glycogen synthase, GYS1-R309H (15). Glycogen synthase is the enzyme responsible for the synthesis of the linear portions of the glycogen molecule. The genetic variant of glycogen synthase associated with PSSM constitutively actives glycogen synthase (16). This means that glycogen metabolism is no longer subject to normal regulation. Glycogen is synthesized even under conditions of energy demand.
The presence of enlarged glycogen granules in muscle tissue of horses with exercise intolerance is not completely correlated with the presence of the genetic variant of glycogen synthase that constitutively activates glycogen synthase (17). In Quarter Horses showing enlarged glycogen granules, 95.6% have one or two copies of the GYS1-R309H variant, while 4.5% have two normal copies of the glycogen synthase gene. In Quarter Horses showing glycogen granules of normal size, 77.7% have two normal copies of the glycogen synthase gene, while 22.4% have one or two copies of the GYS1-R309H variant.
This suggests that genetic variants affecting other genes may cause glycogen storage disease in horses. On this basis, horses with the GYS1-R309H variant are said to have Polysaccharide Storage Myopathy type 1 (PSSM1), while horses showing enlarged glycogen granules but lacking the GYS1-R309H variant are unexplained. In the years since the discovery of the GYS1-R309H variant and its association with PSSM1, no variants of the genes shown in the table above associated with glycogen storage disease in human patients have been found.
One of the additional findings in horses with exercise intolerance is the occurrence of clusters of glycogen granules of normal size, suggesting a different disorder in which glycogen synthesis is constitutively activated. This condition was described as PSSM2 (18). Again, in the years since the discovery of the GYS1-R309H variant and its association with PSSM1, no variants of the equine orthologs of the genes shown in the table above, associated with glycogen storage disease in human patients, have been found.
Arabian horses initially diagnosed with PSSM2 were found to have defects in muscle similar to those found in human patients with a different disease state called Myofibrillar Myopathy. Human patients with Myofibrillar Myopathy have genetic variants of a growing group of genes that affect the structural integrity of muscle. Arabian horses initially diagnosed with PSSM2 do not synthesize abnormal quantities of glycogen; rather, the pooling of glycogen granules of normal size in structurally disorganized myofibrils presents the appearance of a glycogen storage disease (19).
It remains an open question whether there are any genetic variants associated with glycogen storage disease in horses besides GYS1-R309H, associated with PSSM1. For this reason, researchers have suggested an alternative nomenclature in which horses with observable structural defects in muscle and glycogen granules of normal size have Muscle Integrity Myopathy (MIM).
References
1. Lei, KJ, et al. (1994) Identification of mutations in the gene for glucose-6-phosphatase, the enzyme deficient in glycogen storage disease type 1a. J Clin Invest. 93(5): 1994-1999. PubMed ID: 8182131
2. Martiniuk, F, et al (1990) Identification of the base-pair substitution responsible for a human acid alpha glucosidase allele with lower 'affinity' for glycogen (GAA 2) and transient gene expression in deficient cells. Am. J. Hum. Genet. 47: 440-445. PubMed ID: 2203258
3. Shen, J, et al. (1996) Mutations in exon 3 of the glycogen debranching enzyme gene are associated with glycogen storage disease type III that is differentially expressed in liver and muscle. J. Clin. Invest. 98: 352-357. PubMed ID: 8755644
4. Bao, Y, et al. (1996) Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J. Clin. Invest. 97: 941-948. PubMed ID: 8613547
5. Lebo, RV, et al. (1990) Rare McArdle disease locus polymorphic site on 11q13 contains CpG sequence. Hum. Genet. 86: 17-24. PubMed ID: 1701414
6. Burwinkel, B, et al. (1998) Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI (Hers disease). Am. J. Hum. Genet. 62: 785-791. PubMed ID: 9529348
7. Nakajima, H, et al. (1990) Genetic defect in muscle phosphofructokinase deficiency: abnormal splicing of the muscle phosphofructokinase gene due to a point mutation at the 5-prime-splice site. J. Biol. Chem. 265: 9392-9395. PubMed ID: 2140573
8. Hendrickx, J, et al. (1995) Mutations in the phosphorylase kinase gene PHKA2 are responsible for X-linked liver glycogen storage disease. Hum. Molec. Genet. 4: 77-83. PubMed ID: 7711737
9. Tsujino, S, et al. (1993) The molecular genetic basis of muscle phosphoglycerate mutase (PGAM) deficiency. Am. J. Hum. Genet. 52: 472-477. PubMed ID: 8447317
10. Maekawa, M, et al. (1990) Molecular characterization of genetic mutation in human lactate dehydrogenase-A (M) deficiency. Biochem. Biophys. Res. Commun. 168: 677-682. PubMed ID: 2334430
11. Kreuder, J, et al.(1996) Brief report: inherited metabolic myopathy and hemolysis due to a mutation in aldolase A. New Eng. J. Med. 334: 1100-1104. PubMed ID: 8598869
12. Comi, GP, et al. (2001) Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Ann. Neurol. 50: 202-207. PubMed ID: 11506403
13. Ferrell, RE, et al. (1984) Erythrocyte phosphoglucomutase: a family study of a PGM1 deficient allele. Hum. Genet. 67: 306-308. PubMed ID: 6236143
14. Moslemi, A-R, (2010) Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. New Eng. J. Med. 362: 1203-1210. PubMed ID: 20357282
15. McCue, ME, et al. (2008) Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics. 91(5):458-466. PubMed ID: 18358695
16. Maile, CA, et al. (2017) A highly prevalent equine glycogen storage disease is explained by constitutive activation of a mutant glycogen synthase. Biochim Biophys Acta Gen Subj. 1861(1 Pt A):3388-3398. PubMed ID: 27592162
17. McCue, ME et al. (2008) Glycogen synthase 1 (GYS1) mutation in diverse breeds with polysaccharide storage myopathy. J Vet Intern Med. 22(5):1228-1233. PubMed ID: 18691366
18. McCue, ME (2009) Comparative skeletal muscle histopathologic and ultrastructural features in two forms of polysaccharide storage myopathy in horses. Vet Pathol. 46(6):1281-1291. PubMed ID: 19605906
19. Valberg, SJ, et al. (2016) Suspected myofibrillar myopathy in Arabian horses with a history of exertional rhabdomyolysis. Equine Vet J. 48(5):548-56. PubMed ID: 26234161
Share this post
From the blog
The latest industry news, interviews, technologies, and resources.
P4 Allele of MYOZ3
Introduction This blog post explores the P4 allele (S42L) of the equine MYOZ3…
1 Apr 2025
P2 Allele of MYOT
Introduction This blog post explores the P2 allele (S232P) of the equine MYOT…
8 Mar 2025
EquiSeq is a biotech company offering
genetic testing for horses.
Resources
© 2017 – 2023 EquiSeq Inc. All rights reserved.