
Blog
Filamin C (FLNC)
13 Jan 2020

Filamins are a family of actin-binding proteins. In humans and other mammals, there are three filamins: filamin A, filamin B, and filamin C. Filamins A and B are widely expressed, while filamin C expression appears limited to skeletal and cardiac muscle.
The figure below (adapted from [1]) shows the domain structure of filamin C.
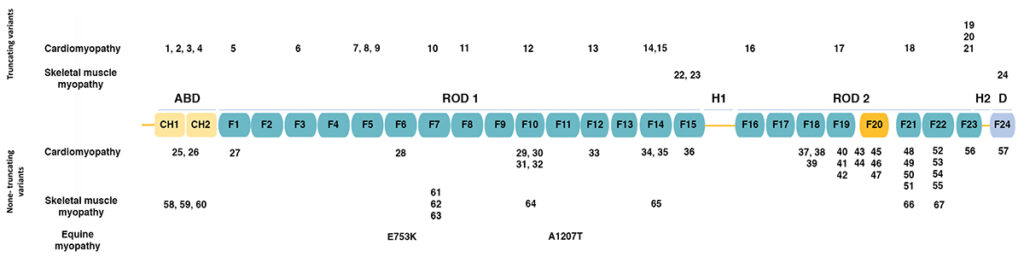
Filamins have an amino-terminal actin-binding domain consisting of two calponin-homology (CH) domains. The rest of the protein consists of 24 immunoglobulin-like filamin domains with two hinge domains. Filamin C differs from filamins A and B in a novel domain 20 that interacts with Z disc proteins, including myotilin and myozenins. While filamin domains 1-15 form a rod, domain 20 is folded over domains 19 and 21 [2]. Filamin domain 24 is required for homodimerization.
The figure also shows the positions of human mutations associated with various types of cardiomyopathy and skeletal muscle myopathy, as summarized in the table below.
| Human FLNC Alleles Associated with Cardiomyopathy | ||||
|---|---|---|---|---|
| Figurea | Alleleb | Domainc | Mutation Typed | Citation |
| 1 | Y7Tfs*51 | ABD | frameshift | 1 |
| 2 | Y83* | ABD | termination | 3 |
| 25 | F106L | ABD | missense | 4 |
| 3 | E108* | ABD | termination | 5 |
| 26 | V123A | ABD | missense | 5 |
| 4 | E238Rfs*14 | ABD | frameshift | 1 |
| 5 | R269* | Filamin 1 | termination | 6 |
| 27 | N290K | Filamin 1 | missense | 5 |
| 6 | R482* | Filamin 3 | termination | 7 |
| 7 | I683Rfs*9 | Filamin 5 | frameshift | 1 |
| 8 | Y705* | Filamin 5 | termination | 3 |
| 9 | Q707* | Filamin 5 | termination | 6 |
| 28 | S792I | Filamin 6 | missense | 8 |
| 10 | Y928* | Filamin 7 | termination | 1 |
| 11 | R991* | Filamin 8 | termination | 7 |
| 29 | A1183L | Filamin 10 | missense | 9 |
| 30 | A1186V | Filamin 10 | missense | 9 |
| 31 | S1194L | Filamin 10 | missense | 1 |
| 12 | V1198Gfs*64 | Filamin 10 | frameshift | 1 |
| 32 | Y1216N | Filamin 10 | missense | 10 |
| 13 | R1370* | Filamin 12 | termination | 3 |
| 33 | G1424V | Filamin 12 | missense | 1 |
| 14 | A1539T | Filamin 14 | termination | 5 |
| 34 | A1551T | Filamin 14 | missense | 8 |
| 15 | L1573* | Filamin 14 | termination | 7 |
| 35 | S1624L | Filamin 14 | missense | 11 |
| 36 | I1666T | Filamin 15 | missense | 1 |
| 16 | G1800* | Filamin 16 | termination | 3 |
| 37 | I1946_T1947dup | Filamin 18 | in-frame duplication | 1 |
| 38 | R1999Q | Filamin 18 | missense | 8 |
| 39 | G2011E | Filamin 18 | missense | 1 |
| 40 | G2039R | Filamin 19 | missense | 12 |
| 17 | Y2100* | Filamin 19 | frameshift duplication | 13 |
| 41 | R2133H | Filamin 19 | missense | 5 |
| 42 | R2140Q | Filamin 19 | missense | 14 |
| 43 | G2151S | Filamin 19/20 | missense | 5 |
| 44 | I2160F | Filamin 19/20 | missense | 11 |
| 45 | V2297M | Filamin 20 | missense | 15 |
| 46 | P2298L | Filamin 20 | missense | 16 |
| 47 | G2299S | Filamin 20 | missense | 1 |
| 48 | H2315N | Filamin 21 | missense | 5 |
| 49 | R2318W | Filamin 21 | missense | 14 |
| 50 | I2359T | Filamin 21 | missense | 1 |
| 18 | Y2373Cfs*7 | Filamin 21 | frameshift | 1 |
| 51 | V2375L | Filamin 21 | missense | 14 |
| 52 | R2410C | Filamin 22 | missense | 1 |
| 53 | E2417P | Filamin 22 | missense | 1 |
| 54 | A2430V | Filamin 22 | missense | 5 |
| 55 | R2495H | Filamin 22 | missense | 1 |
| 19 | P2513Efs*12 | Filamin 23 | frameshift | 17 |
| 20 | Q2549* | Filamin 23 | termination | 1 |
| 21 | C2555* | Filamin 23 | termination | 1 |
| 56 | Y2563C | Filamin 23 | missense | 16 |
| 57 | P2643_L2645del | Filamin 24 | in-frame deletion | 1 |
| Human FLNC Alleles Associated with Skeletal Muscle Myopathy | ||||
| Figurea | Alleleb | Domainc | Mutation Typed | Citation |
| 58 | A193T | ABD | missense | 18 |
| 59 | M222V | ABD | missense | 19 |
| 60 | M251T | ABD | missense | 18 |
| 61 | K899_V904del, insV899_C900 | Filamin 7 | deletion + insertion | 20 |
| 62 | V930_T933del | Filamin 7 | in-frame deletion | 21 |
| 63 | 931_935del | Filamin 7 | in-frame deletion | 22 |
| 64 | Y1216N | Filamin 10 | missense | 10 |
| 65 | A1630V | Filamin 14 | missense | 23 |
| 22 | F1720Lfs*63 | Filamin 15 | frameshift | 24 |
| 23 | G1722Vfs*61 | Filamin 15 | frameshift | 25 |
| 66 | V2375I | Filamin 21 | missense | 26 |
| 67 | T2419M | Filamin 22 | missense | 27 |
| 24 | W2710* | Filamin 24 | termination | 28 |
| Equine FLNC Alleles Associated with Skeletal Muscle Myopathye | ||||
| Figurea | Alleleb | Domainc | Mutation Typed | Citation |
| E753K A1207T | E753K+A1207T (P3) | Filamin 6, 11 | missense | EquiSeq (unpublished) |
aAbbreviation used in Figure.
bAllele nomenclature shows the single-letter code for the wild-type amino acid, the amino acid position in the protein, and the substituted amino acid (for missense alleles) or “*” to indicate a termination codon. Frameshift alleles indicate the identity of the amino acid at the site of the frameshift, the amino acid position, the new amino acid at that position, the designation “fs,” and the number of amino acids before the new termination (*) is reached. For example, E238Rfs*14 means that the glutamic acid (E) at position 238 is now arginine (R), and that there are 14 amino acids from an incorrect reading frame before a termination codon is reached. For more complex alleles (insertions, deletions, etc.), please see the citation for a description.
cDomains in filamin C are as follows: Actin-binding domain (ABD), Filamin repeat (Filamin).
dAllele types are missense (substitution of a different amino acid at that position), termination (mutation of an amino acid codon to a termination codon), frameshift, or more complex types like insertions and deletions.
eThe equine protein model differs from the human protein model, so that equine E753K corresponds to human E793K (Filamin 6 domain) and equine A1207T corresponds to human A1247T (Filamin 11 domain).
Mutations in FLNC may be categorized as truncating (termination codons and frameshift alleles leading to truncation) or non-truncating (missense alleles and in-frame duplications and deletions). Truncating mutations associated with cardiomyopathy are distributed across the length of the protein, while truncating mutations associated with skeletal muscle myopathy occur only in filamin domains 15 and 24. Non-truncating mutations associated with cardiomyopathy and skeletal muscle myopathy are both distributed across the length of the protein.
Some human patients initially identified as having skeletal muscle myopathy also develop cardiomyopathy [16].
A targeted mutation of FLNC in mice, Flnctm1.1Rsdf, is a gene truncation lacking Ig-like repeats 20 through 24 [29]. The Flnctm1.1Rsdf allele destabilizes Flnc mRNA and produces only a trace amount of truncated FLNC protein that is likely inactive. Mice homozygous for Flnctm1.1Rsdf die within minutes of birth from respiratory insufficiency and show multiple defects in skeletal muscle tissue. These include decreased muscle mass, centrally located nuclei, and infiltration of connective tissue, showing that the FLNC protein is essential for normal muscle development [29].
The 24th filamin domain is required for dimerization. A well-studied human allele, W2710*, has a mutation to a termination codon in the 24th Ig-like domain. This mutation is associated with Myofibrillar Myopathy 5 (MFM5) in humans [28]. The mutant protein is unable to dimerize, but nevertheless localizes to the Z disc [30]. In addition, ectopic aggregates of Z disc proteins, including filamin, myotilin, and desmin, form within myofibrils [28, 30]. Electron microscopy shows ultrastructural defects including breaks in the Z disc and Z disc streaming [28, 30].
The table also shows an equine allele nicknamed P3 that bears two missense mutations: E753K, corresponding to human E793K (Filamin 6 domain) and A1270T, corresponding to human A1247T (Filamin 11 domain). These two missense mutations are a haplotype discovered in a horse diagnosed with Myofibrillar Myopathy (MFM) through the identification of desmin-positive inclusions observed in muscle tissue. The two missense mutations are a haplotype in that when one is detected, the other is also detected, in samples of hundreds of horses. P3/P3 homozygotes have been observed and appear to be more severely affected; there is no evidence of cardiomyopathy (EquiSeq, unpublished).
Evidence that the equine P3 allele is pathogenic will be presented in a subsequent blog post.
Citations
[1] Ader F et al. (2019). “FLNC pathogenic variants in patients with cardiomyopathies: Prevalence and genotype-phenotype correlations.” Clin Genet. 96(4): 317-329. PMID: 31245841.
[2] Sutherland-Smith AJ (2011). “Filamin structure, function and mechanics: are altered filamin-mediated force responses associated with human disease?” Biophys Rev. 3:15–23. PMID: 28510233.
[3] Hall CL et al. (2019). “Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype.” Int J Cardiol. pii: S0167-5273(19)33019-0. PMID: 31627847.
[4] Reinstein E et al. (2016). “Congenital dilated cardiomyopathy caused by biallelic mutations in Filamin C.” Eur J Hum Genet. 24(12):1792-1796. PMID: 27601210.
[5] Valdés-Mas R et al. (2014). “Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy.” Nat Commun. 5:5326. PMID: 25351925.
[6] Begay RL et al. (2018). “Filamin C truncation mutations are associated With arrhythmogenic dilated cardiomyopathy and changes in the cell–cell adhesion structures.” JACC Clin Electrophysiol. 4(4):504-514. PMID: 30067491.
[7] Augusto JB et al. (2019). “Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study.” Eur Heart J Cardiovasc Imaging. 2019 Jul 16. pii: jez188. PMID: 31317183.
[8] Jaafar N et al. (2016). “Spectrum of Mutations in Hypertrophic Cardiomyopathy Genes Among Tunisian Patients.” Genet Test Mol Biomarkers. 20(11):674-679. PMID: 27574918.
[9] Kiselev A et al. (2018). “De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy.” Hum Mutat. 39(9):1161-1172. PMID: 29858533.
[10] Avila-Smirnov D et al. (2010). “A novel missense FLNC mutation causes arrhythmia and late onset myofibrillar myopathy with particular histopathology features.” Neuromuscul Disord. 20:623–624. PMID: 22806379.
[11] Brodehl A et al. (2016). “Mutations in FLNC are associated with familial restrictive cardiomyopathy.” Hum Mutat. 37: 269–279. PMID: 26666891.
[12] Cui H et al. (2018). “Mutation profile of FLNC gene and its prognostic relevance in patients with hypertrophic cardiomyopathy.” Mol Genet Genomic Med. 6(6):1104-1113. PMID: 30411535.
[13] Rojnueangnit K et al. (2019). “Identification of gene mutations in primary pediatric cardiomyopathy by whole exome sequencing.” Pediatr Cardiol. 2019 Nov 11. [Epub ahead of print] PMID: 31712860.
[14] Gómez J et al. (2017). “Screening of the filamin C gene in a la rge cohort of hypertrophic cardiomyopathy patients.” Circ Cardiovasc Genet. 10(6). pii: e001584. PMID: 28356264.
[15] Tucker NR et al. (2017). “Novel mutation in FLNC (Filamin C) causes familial restrictive cardiomyopathy.” Circ Cardiovasc Genet. 10(6). pii: e001780. PMID: 29212899.
[16] Schubert J et al. (2018). “Novel pathogenic variants in filamin C identified in pediatric restrictive cardiomyopathy.” Hum Mutat. 39(12):2083-2096. PMID: 30260051.
[17] Mangum KD and Ferns SJ (2018). “A novel familial truncating mutation in the filamin C gene associated with T cardiac arrhythmias.” Eur J Med Genet. 62(4):282-285. PMID: 30118858.
[18] Duff RM et al. (2011). “Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy.” Am J Hum Genet. 88(6):729-740. PMID: 21620354.
[19] Gemelli C et al. (2019). “A novel mutation in the N-terminal acting-binding domain of Filamin C protein causing a distal myofibrillar myopathy.” J Neurol Sci. 398:75-78. PMID: 30685713.
[20] Luan X et al. (2010). “A novel heterozygous deletion-insertion mutation (2695–2712 del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy in a large Chinese family.” Neuromuscul Disord. 20:390–396. PMID: 20417099.
[21] Shatunov A et al. (2009). “In-frame deletion in the seventh immunoglobulin-like repeat of filamin C in a family with myofibrillar myopathy.” Eur J Hum Genet. 17(5):656-663. PMID: 19050726.
[22] Miao J et al. (2018). “A case report: a heterozygous deletion (2791_2805 del) in exon 18 of the filamin C gene causing filamin C-related myofibrillar myopathies in a Chinese family.” BMC Neurol. 18(1):79. PMID: 29866061.
[23] Özyilmaz B et al. (2019). “Impact of next-generation sequencing panels in the evaluation of limb-girdle muscular dystrophies.” Ann Hum Genet. 83(5):331-347. PMID: 31066050.
[24] Guergueltcheva V et al. (2011). “Distal myopathy with upper limb predominance caused by filamin C haploinsufficiency.” Neurology. 77(24):2105-14. PMID: 22131542.
[25] Rossi D et al. (2017). “A novel FLNC frameshift and an OBSCN variant in a family with distal muscular dystrophy.” PLoS One. 12(10):e0186642. PMID: 29073160.
[26] Chen J et al. (2019). “A mutation in the filamin c gene causes myofibrillar myopathy with lower motor neuron syndrome: a case report.” BMC Neurol. 19(1):198. PMID: 31421687.
[27] Tasca G et al. (2012). “Novel FLNC mutation in a patient with myofibrillar myopathy in combination with late-onset cerebellar ataxia.” Muscle Nerve. 46:275–282. PMID: 22806379.
[28] Vorgerd M et al. (2005). “A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy.” Am J Hum Genet. 77(2):297-304. PMID: 15929027.
[29] Dalkilic I et al. (2006). “Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure.” Mol Cell Biol. 26(17):6522-34. PMID: 16914736.
[30] Chevessier F et al. (2015). “Myofibrillar instability exacerbated by acute exercise in filaminopathy.” Hum Mol Genet. 24(25):7207-7220. PMID: 26472074.
Share this post
From the blog
The latest industry news, interviews, technologies, and resources.
P4 Allele of MYOZ3
Introduction This blog post explores the P4 allele (S42L) of the equine MYOZ3…
1 Apr 2025
P2 Allele of MYOT
Introduction This blog post explores the P2 allele (S232P) of the equine MYOT…
8 Mar 2025
EquiSeq is a biotech company offering
genetic testing for horses.
Resources
© 2017 – 2023 EquiSeq Inc. All rights reserved.